




Hara T, Hashimoto Y, Akuzawa T, Hirai R, Kobayashi H, Sato K (IMCR, Gunma Univ.)
About
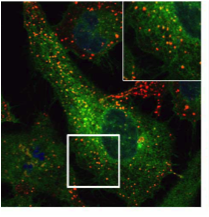
Peripheral myelin protein 22 (PMP22) resides in the plasma membrane and is required for myelin formation in the peripheral nervous system. Many PMP22 mutants accumulate in excess in the endoplasmic reticulum (ER) and lead to the inherited neuropathies of Charcot-Marie-Tooth (CMT) disease. However, the mechanism through which PMP22 mutants accumulate in the ER is unknown. Here, we studied the quality control mechanisms for the PMP22 mutants L16P and G150D, which were originally identified in mice and patients with CMT. We found that the ER-localised ubiquitin ligase Hrd1/SYVN1 mediates ER-associated degradation (ERAD) of PMP22(L16P) and PMP22(G150D), and another ubiquitin ligase, gp78/AMFR, mediates ERAD of PMP22(G150D) as well. We also found that PMP22(L16P), but not PMP22(G150D), is partly released from the ER by loss of Rer1, which is a Golgi-localised sorting receptor for ER retrieval. Rer1 interacts with the wild-type and mutant forms of PMP22. Interestingly, release of PMP22(L16P) from the ER was more prominent with simultaneous knockdown of Rer1 and the ER-localised chaperone calnexin than with the knockdown of each gene. These results suggest that CMT disease-related PMP22(L16P) is trapped in the ER by calnexin-dependent ER retention and Rer1-mediated early Golgi retrieval systems and partly degraded by the Hrd1-mediated ERAD system.
Paper information
Rer1 and calnexin regulate endoplasmic reticulum retention of a peripheral myelin protein 22 mutant that causes type 1A Charcot-Marie-Tooth disease.
Hara T, Hashimoto Y, Akuzawa T, Hirai R, Kobayashi H, Sato K*
Sci Rep. 11;4:6992. 2014
Online URL
http://www.ncbi.nlm.nih.gov/pubmed/25385046