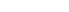





Ryota Inoue 1,#, Kazuki Tajima 2,#, Tomoko Okuyama 2, Chisato Sakai 1, Tamaki Kanno 1, Meng Zou 1, Mayu Kyohara 2, Kohichi Matsunaga 1, Takahiro Tsuno 1, Junyu Luo 1, Emi Ishida 1, Shin-ichi Horike 3, Tohko Hirano 4, Noriaki Arakawa 5, Tatsuya Kin 6, A.M. James Shapiro 6, Yasuo Terauchi 2, Hideru Obinata4, Hisashi Hirano 1,7, and Jun Shirakawa 1,2,* (1. Laboratory of Diabetes and Metabolic Disorders, Institute for Molecular and Cellular Regulation (IMCR), Gunma University, Japan; 2. Department of Endocrinology and Metabolism, Graduate School of Medicine, Yokohama City University, Japan; 3. Division of Integrated Omics Research, Research Center for Experimental Modeling of Human Disease, Kanazawa University, Kanazawa, Japan; 4. Core Facility Management and Technical Collaboration Center, Gunma University, Maebashi, Japan; 5. Division of Medicinal Safety Science, National Institute of Health Sciences, Kawasaki, Japan; 6. Clinical Islet Laboratory and Clinical Islet Transplant Program, University of Alberta, Edmonton, Canada; 7. Advanced Medical Research Center, Yokohama City University, Yokohama, Japan; #: equal contributions, *Corresponding Author)
About
Endoplasmic reticulum (ER) stress is a critical driver of pancreatic β-cell dysfunction and apoptosis. Although metformin, a drug used to treat type 2 diabetes, primarily decreases blood glucose levels by improving insulin sensitivity, its direct effects on β-cell survival remain unclear. Here, we investigated the effect of metformin on β-cell stress responses under ER stress conditions. Thapsigargin (Tg)-induced ER stress increased β-cell apoptosis in mouse islets, which was prevented by metformin in a dose-dependent manner. Treatment with metformin for 24 h suppressed the Tg-induced upregulation of unfolded protein response (UPR)-related genes, as confirmed by transcriptomic and pathway analyses. Quantitative proteomics revealed that Tg inhibited eIF2 signaling and protein translation, both of which were partially restored by metformin. Enrichment analysis further indicated the attenuation of apoptotic pathways in metformin-treated islets. Polysome profiling and puromycin incorporation assays demonstrated that metformin reduced protein translation independently of ER stress. Metformin promoted the dephosphorylation of 4E-BP1, a key initiator of cap-dependent protein translation that is activated by phosphorylation, and the antiapoptotic effect of metformin was abolished by 4E-BP1 knockdown in MIN6 cells. Taken together, these findings reveal a cytoprotective mechanism of metformin in β-cells, in which metformin suppresses ER stress-induced apoptosis through 4E-BP1-mediated inhibition of mRNA translation. This study highlights a β-cell-intrinsic action of metformin that may contribute to its long-term therapeutic benefits in diabetes management.
Paper information
Ryota Inoue, Kazuki Tajima, Tomoko Okuyama, Chisato Sakai, Tamaki Kanno, Meng Zou, Mayu Kyohara, Kohichi Matsunaga, Takahiro Tsuno, Junyu Luo, Emi Ishida, Shin-ichi Horike, Tohko Hirano, Noriaki Arakawa, Tatsuya Kin, A.M. James Shapiro, Yasuo Terauchi, Hideru Obinata, Hisashi Hirano, and Jun Shirakawa. Metformin suppresses β-cell apoptosis under ER stress by inhibiting protein translation. Metabolism. in press, 156607, 2026. doi: 10.1016/j.metabol.2026.156607
Online URL
https://www.sciencedirect.com/science/article/pii/S0026049526001174